How to create a custom structure visualization
- Daniël Brouwer (Unlicensed)
- Sergio Maduro
The basic steps to use the "visualize data in structure module" are:
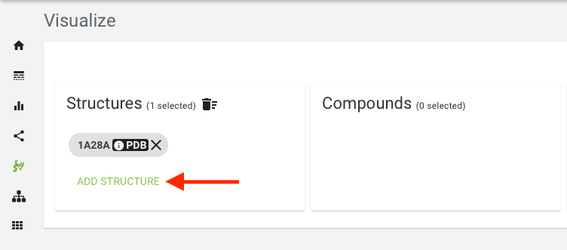
- Select one or more structure files from the "Structures" table by clicking "Add structure" button. (The "System default template" is by default selected. Users can change this by clicking the settings icon.)

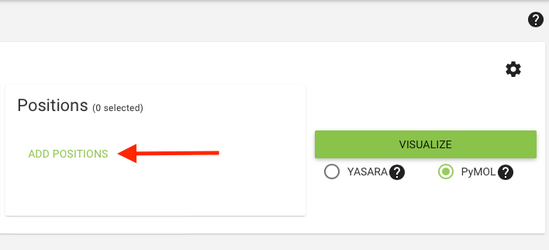
2. Select or deselect position data (custom, hotspot, correlated mutations, conservation, etc) that will be visualized in the selected structures by clicking the "Add positions" button.
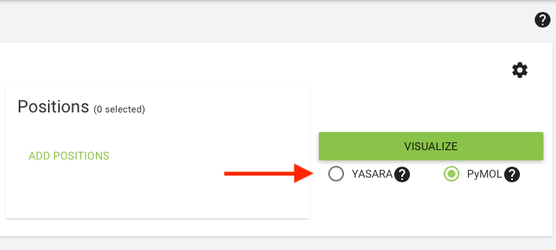
3. Users can select their preferred visualization tool by clicking the YASARA / PyMol radio buttons. Download the visualization file by clicking the "Visualize" button.
In the structures table different structures can be selected. ALL STRUCTURES CAN BE COMBINED because all structures are superimposed INCLUDING the co-crystalized compound (e.g inhibitors, substrate analogs, etc).
Structures types are as followed:
- Templates: The templates of the alignments.
- PDB files in alignment: PDB structures aligned in one of the subfamilies.
- Non-aligned pdb files: PDB structures that could be superimposed, but were not included in the alignment.*
- Models: Models that have been generated.**
Structure compounds can also be included by clicking on the "Include compounds" checkmark next to the search box or be manually selected/deselected under the "Select compounds" table column.

* Why 3DM did not include PDB files in the alignment can have multiple reasons, but mostly this is because the structural similarity is low compared to the other family members.
** To generate models, go to the protein page of the sequence for which a model needs to be generated and click "generate model".